Anemia Mediterranea (Morbo di Cooley)
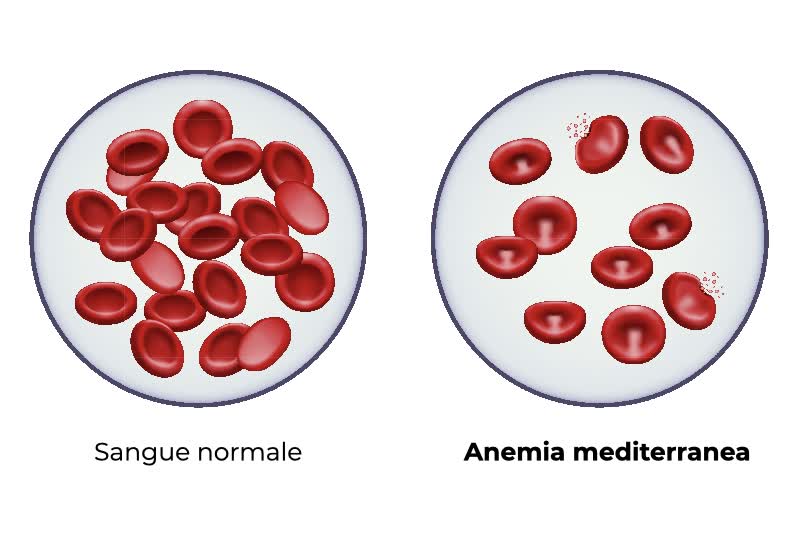
L’anemia mediterranea, detta anche beta-talassemia major o morbo di Cooley, è una forma di talassemia, un’anemia ereditaria causata da mutazioni in un gene necessario per la sintesi dell’emoglobina. I pazienti affetti da anemia mediterranea non producono una delle subunità proteiche dell’emoglobina. La sintesi dell’emoglobina non avviene correttamente e di conseguenza si ha una grave forma di anemia.
Senza emotrasfusioni regolari l’anemia mediterranea risulta letale. I pazienti con anemia mediterranea, anche con una terapia trasfusionale adeguata, hanno un’aspettativa di vita ridotta a causa delle conseguenze della malattia e dei trattamenti.
- 1 Quali sono le cause dell’anemia mediterranea?
- 2 Quali sono i sintomi dell’anemia mediterranea?
- 3 Aspettative di vita
- 4 Come si effettua la diagnosi di anemia mediterranea?
- 5 Come si cura l’anemia mediterranea?
- 6 Quali sono le possibili conseguenze dell’anemia mediterranea?
- 7 Fonti
Quali sono le cause dell’anemia mediterranea?
Quali sono i sintomi dell’anemia mediterranea?
L’anemia mediterranea si manifesta tra i 6 e i 24 mesi di vita. I bambini mostrano un ritardo di crescita e hanno la cute pallida; possono avere problemi di alimentazione, diarrea, irritabilità, attacchi di febbre ricorrenti e ingrandimento progressivo dell’addome causato dall’ingrossamento della milza (splenomegalia), l’organo che elimina i globuli rossi.
I pazienti con anemia mediterranea che non hanno accesso a una terapia trasfusionale ottimale, presentano cute pallida, ittero, modificazioni dello scheletro.
Alcuni esempio possono essere gli zigomi prominenti, ma anche ingrossamento del fegato, della milza (epatosplenomegalia), e ulcere delle gambe. I pazienti possono avere un’eccessiva coagulazione del sangue (ipercoagulopatia) che predispone alla trombosi e ad altri eventi cardiovascolari, e vanno incontro ad osteoporosi già in giovane età.
Aspettative di vita
Come si effettua la diagnosi di anemia mediterranea?
Quando l’esame oggettivo e la storia personale e famigliare fanno sospettare che il paziente sia affetto da anemia mediterranea, si eseguono l’emocromo, che valuta numerosi parametri relativi ai globuli rossi, e l’analisi qualitativa e quantitativa dell’emoglobina, mediante elettroforesi o HPLC (cromatografia liquida ad alta prestazione). Vengono poi eseguiti degli esami ematochimici, tra cui l’analisi dei livelli di ferro, ferritina, transferrina e bilirubina.
Utilizzando test genetici si identificano le mutazioni nel gene che codifica per la catena beta. È possibile eseguire la diagnosi prenatale di anemia mediterranea mediante amniocentesi o villocentesi.
Un esame genetico preconcezionale consente a chi potrebbe avere ereditato delle mutazioni di sapere se è portatore o meno della malattia. Due individui portatori sani di beta-talassemia hanno, ad ogni concepimento, il 25% di possibilità di generare un figlio con talassemia major, il 50% di possibilità di generare un portatore sano e il 25% di possibilità di generare un figlio senza difetto genetico.
Come si cura l’anemia mediterranea?
L’anemia mediterranea è una talassemia trasfusione-dipendente: per sopravvivere i pazienti hanno bisogno della terapia trasfusionale, che consiste nel sottoporsi a emotrasfusioni regolari per tutta la vita.
Le trasfusioni di sangue causano un accumulo di ferro nel corpo, che danneggia organi vitali come il fegato e il cuore, e di conseguenza il paziente deve assumere una terapia ferrochelante a base di farmaci che legano il ferro in eccesso e ne permettono l’eliminazione.
Il trapianto di midollo o il trapianto di cellule staminali ematopoietiche da donatore è l’unico trattamento che permette di curare definitivamente la malattia.
Quali sono le possibili conseguenze dell’anemia mediterranea?
- Stimolazione eccessiva del midollo osseo: Il nostro corpo cerca di compensare la mancanza di globuli rossi sani producendone di più. Questo sforzo extra può causare un’espansione del midollo osseo, che a sua volta può portare a deformazioni ossee e altri problemi strutturali.
- Produzione inefficace di globuli rossi: Anche se il corpo cerca di produrne di più, i globuli rossi creati non sono funzionali come dovrebbero essere, il che significa che non possono trasportare ossigeno in modo efficiente. Questo porta a una condizione nota come anemia.
- Accumulo di ferro dovuto alle trasfusioni: I pazienti con anemia mediterranea spesso necessitano di trasfusioni di sangue per aumentare il numero di globuli rossi. Tuttavia, queste trasfusioni possono portare a un eccesso di ferro nel corpo, che può danneggiare organi vitali come il fegato, il cuore e le ghiandole che producono ormoni.
- Nei bambini e adolescenti, può verificarsi un ritardo nella crescita e nello sviluppo sessuale, a causa sia dell’anemia che dell’accumulo di ferro.
- Gli adulti possono sviluppare problemi più gravi, come:
- Cirrosi epatica: Danni al fegato a causa dell’accumulo di ferro.
- Problemi endocrini: Disturbi come il diabete e l’ipogonadismo (una condizione che impatta la produzione di ormoni sessuali) possono emergere a causa dell’eccesso di ferro che interferisce con le ghiandole endocrine.
- Problemi cardiaci: L’accumulo di ferro nel cuore può portare a condizioni come la cardiomiopatia dilatativa (un tipo di malattia cardiaca che influisce sulla capacità del cuore di pompare sangue) e aritmie (battiti cardiaci irregolari).
- Trombosi venosa e infezioni: L’eccesso di ferro e l’anemia possono aumentare il rischio di coaguli di sangue e di infezioni.
- Osteoporosi: Una riduzione della densità ossea, che può portare a fratture più facilmente.
Fonti
- Taher AT, et al. β-Thalassemias. N Engl J Med 2021;384:727-743. doi:10.1056/NEJMra2021838
- Origa R. β-thalassemia. Genet Med. 2017;19(6):609-619. doi:10.1038/gim.2016.173
- Muncie HL, Campbell JS. Alpha and Beta Thalassemia. Am Fam Physician. 2009;80(4):339-444.
- Fondazione Theleton – Talassemie