Sindrome del QT lungo (LQTS)
La sindrome del QT lungo è una malattia cardiaca che può causare la morte improvvisa. Può essere genetica o insorgere come conseguenza di disordini elettrolitici o terapie farmacologiche. Viene evidenziata mediante elettrocardiogramma. Si interviene farmacologicamente, mediante defibrillatore cardiaco impiantabile, chirurgicamente o, ove possibile, rimuovendo le cause.
- 1 Patologie correlate alla sindrome del QT lungo
- 2 Sintomi e caratteristiche della sindrome del QT lungo
- 3 Come si diagnostica la sindrome del QT lungo
- 4 Come si cura la sindrome del QT lungo
- 5 Fonti
Patologie correlate alla sindrome del QT lungo
La sindrome del QT lungo (LQTS, dall’inglese Long QT Syndrome) è una malattia cardiaca caratterizzata da aritmie ventricolari gravi che possono portare alla sincope o alla morte cardiaca improvvisa. La morte cardiaca improvvisa è un evento che colpisce persone giovani e altrimenti sane: è una delle più frequenti cause di morte improvvisa nei giovani atleti. Dipende spesso da una malattia aritmogena, una patologia che coinvolge i meccanismi che controllano il ritmo cardiaco.
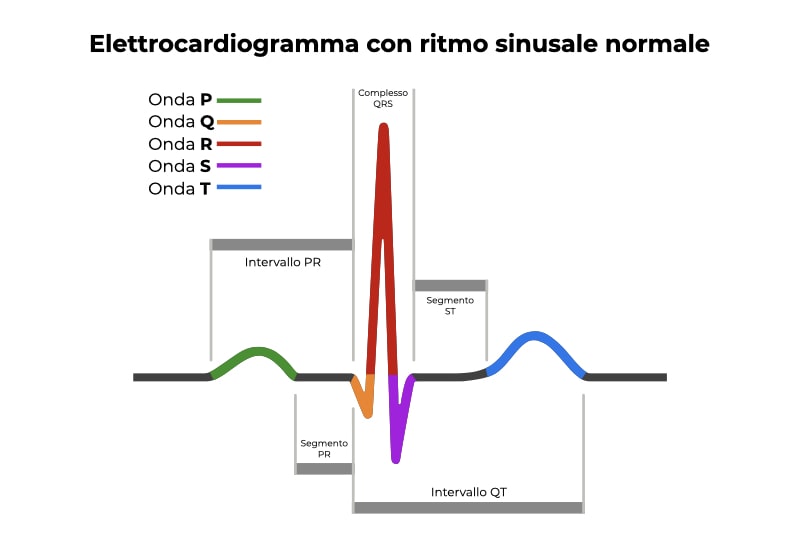
Le malattie aritmogene congenite che sono state associate alla morte cardiaca improvvisa includono la sindrome di Brugada, la tachicardia ventricolare polimorfa catecolaminergica (CPVT), la sindrome del QT corto (SQTS; Short QT Syndrome) e, appunto, la sindrome del QT lungo. Con il termine “intervallo QT” ci si riferisce al tracciato dell’elettrocardiogramma.
L’elettrocardiogramma consente di visualizzare la depolarizzazione e la ripolarizzazione degli atri e dei ventricoli del cuore sotto forma di onde (onde P, Q, R, S e T). L’intervallo QT rappresenta l’intervallo di tempo che intercorre tra l’inizio della depolarizzazione dei ventricoli e la fine della ripolarizzazione dei ventricoli.
L’elemento caratteristico della sindrome del QT lungo è un intervallo QT superiore a 480 millisecondi. La sindrome del QT lungo può essere congenita o acquisita. Sono state descritte diverse varianti genetiche di questa patologia, indicate con la sigla LQT seguita da un numero, le più comuni sono le forme LQT1, LQT2 e LQT3, che complessivamente costituiscono il 90% dei casi.
Sintomi e caratteristiche della sindrome del QT lungo
La sindrome si manifesta con alcuni sintomi caratteristici come:
- tachiaritmie (il battito è irregolare e accelerato);
- palpitazioni;
- vertigini;
- convulsioni;
- sincope; o
- arresto cardiaco.
Nella sindrome LQT1 le sincopi o l’arresto cardiaco si verificano tipicamente durante uno sforzo (il nuoto risulta particolarmente pericoloso); i pazienti affetti da una forma particolare di sindrome LQT1, la sindrome di Jervell-Lange Nielsen, soffrono di sordità. Nella sindrome LQT2 le aritmie o la sincope sono generalmente scatenate da stress acuto, spesso causati da uno stimolo uditivo come il suono di una sveglia. Nella sindrome LQT3 le aritmie si verificano più spesso nel sonno o durante il riposo.
Il rischio di morte improvvisa è più alto per i pazienti con un intervallo QT superiore a 500 millisecondi, per i pazienti con le forme LQT2, LQT3 o varianti rare come la sindrome di Jervell-Lang Nielsen o la sindrome di Timothy (LQT8) e per quelli in cui le aritmie insorgono in età giovanile.
Come si diagnostica la sindrome del QT lungo
La diagnosi si basa sui sintomi, sulla storia familiare del paziente e sull’elettrocardiogramma. Gli esami ematochimici possono rivelare uno squilibrio degli elettroliti. Il medico raccoglie informazioni sulla presentazione dei sintomi e sulla presenza in famiglia di casi di sincope o morte improvvisa in giovane età. La sindrome del QT lungo andrebbe sempre sospettata in un paziente giovane che ha episodi sincopali, soprattutto se ci sono stati casi di morte cardiaca improvvisa in famiglia. Il test del DNA consente di confermare la diagnosi o di diagnosticare una forma congenita nei casi dubbi.
Come si cura la sindrome del QT lungo
La terapia farmacologica per i pazienti con LQTS congenita consiste nella somministrazione di un beta-bloccante. Nei pazienti in cui i sintomi si presentano anche con la terapia e in quelli ad alto rischio di tachiaritmie gravi e morte improvvisa viene solitamente impiantato un defibrillatore cardiaco impiantabile (ICD, Implantable Cardioverter Defibrillator), un dispositivo che rileva l’aritmia potenzialmente fatale ed eroga uno shock elettrico per riportare il ritmo cardiaco alla normalità.
In casi selezionati si ricorre alla denervazione simpatica sinistra, in cui si rimuovono alcune terminazioni del sistema nervoso simpatico che modulano la frequenza cardiaca. Nei pazienti con LQTS acquisita il trattamento consiste nell’eliminazione della causa.
Fonti
- Kramer DB, Zimetbaum PJ. Long-QT Syndrome. Cardiol Rev. 2011;19(5):217-225. doi:10.1097/CRD.0b013e3182203504;
- Schwartz PJ, Crotti Lia, et al. Long-QT syndrome: from genetics to management. Circ Arrhythm Electrophysiol. 2012;5(4):868-877. doi:10.1161/CIRCEP.111.962019;
- Shah SR, Park K. Long QT Syndrome: a comprehensive review of the literature and current evidence. Curr Probl Cardiol. 2019;44(3):92-106. doi:10.1016/j.cpcardiol.2018.04.002