Fibrosi cistica
La fibrosi cistica è una malattia ereditaria dovuta a mutazioni nel gene CFTR. Danneggia la funzionalità respiratoria e causa problemi nutrizionali. L’esame diagnostico specifico è il test del sudore. Non può essere curata, ma, grazie alle strategie di trattamento oggi disponibili, la maggior parte dei pazienti raggiunge l’età adulta.
Descrizione della fibrosi cistica
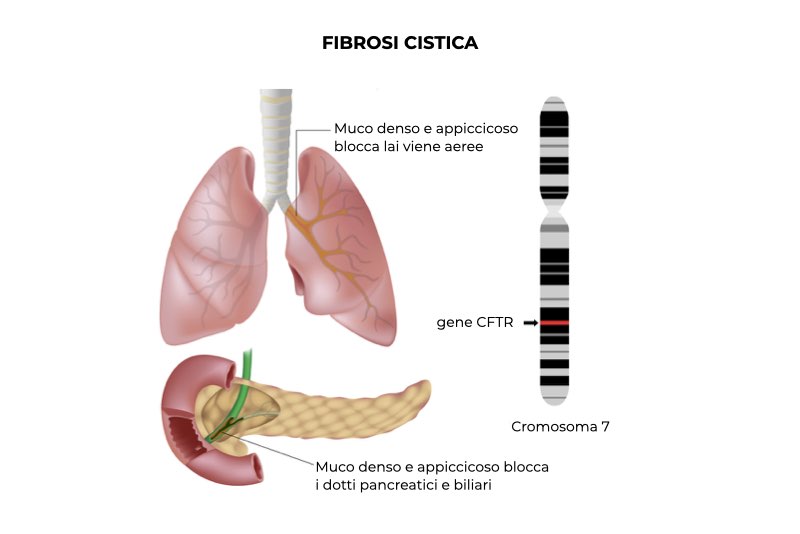
La fibrosi cistica è una malattia genetica causata da mutazioni nel gene CFTR (Cystic Fibrosis Transmembrane Regulator) che codifica per una proteina (CFTR), il cui compito è trasportare il cloro e il bicarbonato dentro e fuori le cellule epiteliali per mantenere idratata la superficie epiteliale. La fibrosi cistica è una malattia genetica a trasmissione autosomica recessiva: ciò significa che il gene coinvolto si trova su un autosoma (un cromosoma diverso dai cromosomi sessuali) e che la malattia si manifesta solo quando sono presenti due copie mutate del gene, una ereditata dal padre e una dalla madre.
Una coppia di portatori sani ha, ad ogni concepimento, il 25% di probabilità di generare un figlio affetto dalla malattia, il 50% di generare un portatore sano e il 25% di generare un figlio senza mutazioni nel gene CFTR. È la malattia genetica più frequente in Italia.
Sono state identificate oltre 2.000 mutazioni diverse del gene CFTR che hanno come risultato il fatto che la proteina non è sintetizzata, non è processata o regolata correttamente o funziona male. La mutazione più comune, è quella in cui si ha la perdita (delezione) dell’amminoacido fenilalanina nella posizione 508 (indicata con F508del), che fa sì che la proteina non maturi correttamente.
Cause e conseguenze della fibrosi cistica
La fibrosi cistica causa cambiamenti patologici negli organi in cui è espressa la proteina CFTR, tra cui le vie respiratorie, il pancreas, il tratto epatobiliare e l’apparato riproduttore maschile. I danni più rilevanti interessano le vie respiratorie. Il difetto nel trasporto del cloro rende il muco che riveste le vie aeree più denso e le ciglia dell’epitelio bronchiale (piccoli filamenti che si trovano sulla superficie delle cellule epiteliali) fanno fatica a farlo muovere verso la gola (questo meccanismo di pulizia delle vie aeree viene chiamato “clearance mucociliare”)
I batteri introdotti con la respirazione trovano nel muco ristagnante condizioni favorevoli per proliferare e causare infezioni polmonari croniche. Tra le infezioni più comuni ci sono quelle da Pseudomonas aeruginosas, Staphylococcus aureus e Haemophilus influenzae. L’infiammazione delle vie respiratorie danneggia il tessuto polmonare e porta a insufficienza respiratoria.
La maggior parte dei pazienti con fibrosi cistica soffre di insufficienza pancreatica: il pancreas non riversa nell’intestino gli enzimi digestivi che produce e questi si accumulano nei dotti pancreatici, danneggiando l’organo; alcune volte il danno pancreatico provoca il diabete mellito. L’ostruzione delle vie biliari causa infiammazione del fegato e steatosi epatica (accumulo di grasso) che può evolvere in cirrosi epatica; sono comuni i calcoli biliari. La fibrosi cistica causa infertilità maschile: i pazienti producono spermatozoi e hanno una capacità sessuale normale, ma l’assenza congenita dei dotti deferenti, i canali che trasportano lo sperma, impedisce la fecondazione.
Come si presenta la fibrosi cistica
A livello dell’apparato respiratorio i segni e i sintomi della fibrosi cistica includono tosse cronica produttiva (tosse grassa), infezioni, bronchiectasia, pansinusite (una sinusite che coinvolge tutti i seni nasali) e polipi nasali.
La malattia gastrointestinale si manifesta con cattiva digestione, malnutrizione, scarso accrescimento e pancreatite ricorrente. Una manifestazione tipica è l’ileo da meconio: le prime feci del neonato (meconio) risultano molto dense e collose e causano un’occlusione intestinale a livello dell’ileo (l’ultimo tratto dell’intestino tenue) per la cui risoluzione è necessario intervenire chirurgicamente. I pazienti con fibrosi cistica possono andare incontro a una grave disidratazione detta “sindrome Pseudo-Bartter”. Il difetto nel trasporto del cloro a livello dell’epitelio delle ghiandole sudoripare fa sì che il sudore dei pazienti con fibrosi cistica sia 3-5 volte più salato del normale.
Come si diagnostica la fibrosi cistica
Il test più specifico per la diagnosi di fibrosi cistica è il test del sudore che consiste nell’analisi della concentrazione del cloro nel sudore. I test genetici consentono di identificare le mutazioni associate alla malattia. La fibrosi cistica è una delle malattie incluse nel programma di screening neonatale obbligatorio e gratuito per tutti i bambini che nascono in Italia, in cui si analizza una goccia di sangue prelevata dal tallone a due-tre giorni di vita per rilevare condizioni potenzialmente fatali o invalidanti nei neonati, prima che il bambino mostri i segni o i sintomi della malattia.
Come si cura la fibrosi cistica
Ad oggi non esiste una cura per la fibrosi cistica. Nel passato gli individui con fibrosi cistica, chiamata allora mucoviscidosi, morivano prima di raggiungere l’età scolare; grazie al miglioramento delle terapie per controllare i sintomi e prevenire le complicanze l’aspettativa di vita è aumentata e oggi supera i 40 anni.
È essenziale prevenire le infezioni polmonari adottando misure igieniche corrette. In caso di infezione e a scopo di profilassi vengono somministrate alte dosi di farmaci antibiotici. Si utilizzano tecniche per rimuovere il muco bronchiale (drenaggio delle secrezioni) e aerosol a base di mucolitici per fluidificarlo. In caso di insufficienza respiratoria i pazienti ricorrono all’ossigenoterapia, ma, quando la malattia polmonare è molto avanzata, l’unica opzione disponibile è il trapianto di polmone. L’insufficienza pancreatica viene trattata somministrando estratti pancreatici. Si interviene sulla dieta, aumentando l’apporto energetico.
In anni recenti sono stati sviluppati i primi farmaci modulatori della proteina CFTR che ne correggono il difetto di funzionamento (ivacaftor) o la maturazione e l’esposizione sulla superficie delle cellule epiteliali (tezacaftor e lumacaftor).
Fonti
- Ratjen F, Döring G. Cystic fibrosis. Lancet 2003; 361:681-689. doi:10.1016/S0140-6736(03)12567-6;
- De Boek K. Cystic fibrosis in the year 2020: A disease with a new face. Acta paediatrica. 2020;109:893–899. doi:10.1111/apa.15155;
- Fondazione Ricerca Fibrosi Cistica ;
- Osservatorio Screening neonatale https://www.osservatorioscreening.it/lo-screening-neonatale-per-la-fibrosi-cistica/.