Cardiomiopatia dilatativa
La cardiomiopatia dilatativa è una patologia che fa parte della famiglia delle cardiomiopatie determinata da fattori ereditari o acquisiti, che comporta la dilatazione della parete dei ventricoli (prevalentemente quello sinistro) del cuore. Questa condizione può portare a una progressiva disfunzione dell’attività contrattile con conseguente scompenso cardiaco e aritmie che possono determinare morte cardiaca improvvisa.
- 1 Cos’è la cardiomiopatia dilatativa
- 2 La diagnosi della cardiomiopatia dilatativa
- 3 I sintomi e le complicanze della cardiomiopatia dilatativa
- 4 Cura e trattamento della cardiomiopatia dilatativa
- 5 Fonti
Cos’è la cardiomiopatia dilatativa
La cardiomiopatia dilatativa ha una prevalenza stimata di circa 1 caso ogni 250–500 persone ed rappresenta una delle principali cause di trapianto cardiaco nei Paesi occidentali. Circa il 30–50% dei casi di cardiomiopatia dilatativa sono di natura ereditaria, altre possibili cause sono i fattori ambientali, infettivi e sistemici. La cardiomiopatia dilatativa può insorgere a qualsiasi età, ma è più comune nei pazienti di età compresa tra i 40 e i 59 anni.
Il cuore, l’organo responsabile dell’afflusso di sangue in tutti i tessuti del corpo umano, è suddiviso in quattro cavità, separate da strutture anatomiche dette setti: gli atri, destro e sinistro, che accolgono rispettivamente il sangue proveniente dalla circolazione sistemica (che è povero di ossigeno) e polmonare (ricco di ossigeno), e i ventricoli, destro e sinistro, che rispettivamente, contraendosi, spingono il sangue attraverso la circolazione polmonare e sistemica; data la sua natura contrattile, il cuore è costituito da tessuto muscolare, detto tessuto miocardico.
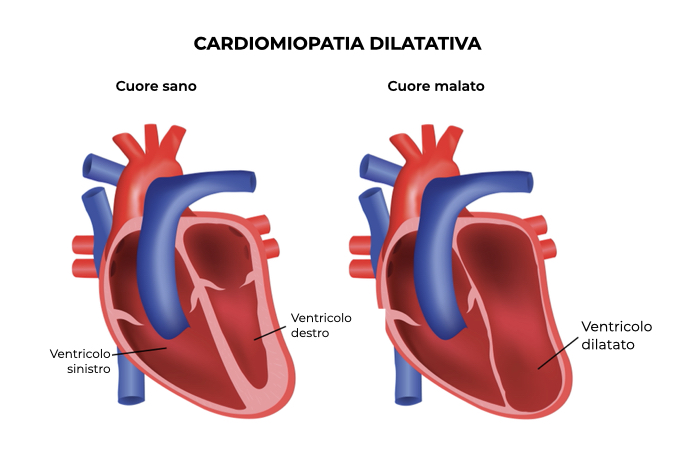
Nella cardiomiopatia dilatativa, a causa di vari fattori, tra cui mutazioni che coinvolgono i geni delle cellule muscolari cardiache, si verifica una dilatazione della parete del ventricolo (spesso sinistro), che porta a fenomeni di rimodellamento: nel corso del tempo il ventricolo assume una forma sferica e diminuisce la sua capacità contrattile. Il risultato è un anormale meccanismo di riempimento del ventricolo, con una riduzione del volume di sangue immesso in circolo e un aumento delle pressioni di riempimento cardiache e riduzione della portata cardiaca: tutti gli adattamenti fisiopatologici indotti dalla cardiomiopatia dilatativa provocano un minor apporto di sangue e al tempo stesso una maggior richiesta di ossigeno da parte dei tessuti.
In base alle cause di insorgenza si può distinguere:
- cardiomiopatia dilatativa ereditaria, dovuta a mutazioni a carico dei geni delle cellule cardiache muscolari, questa può seguire un nella maggior parte dei casi ereditaria segue un pattern autosomico dominante; forme recessive e legate al cromosoma X sono meno comuni (rischio del 25% che il figlio sia malato);
- cardiomiopatia dilatativa acquisita, che può essere a sua volta idiopatica (quando non è possibile identificare una causa specifica), oppure può essere secondaria a cardiopatia ischemica, miocarditi, infezioni virali, esposizione a tossine, abuso di alcol o sostanze stupefacenti, disordini metabolici o endocrini, miocarditi, infezioni virali, esposizione a tossine, alcol e droghe, disfunzioni metaboliche ed endocrine.
La diagnosi della cardiomiopatia dilatativa
La diagnosi di cardiomiopatia dilatativa avviene in seguito ad approfondita visita medica e a diverse pratiche diagnostiche, tra cui:
- elettrocardiogramma (ECG),un esame che traccia l’andamento elettrico del battito cardiaco e può evidenziare aritmie e alterazioni della conduzione elettrica;
- ecocardiogramma, esame di primo livello che consente di valutare dilatazione ventricolare e funzione sistolica e valutare la presenza di anomalie;
- risonanza magnetica cardiaca (cardio RM), esame che si esegue in un macchinario capace di generare un campo magnetico e che consente di fornire ulteriori informazioni sulla struttura del ventricolo dilatato;
- biopsia endomiocardica, indicata solo in casi selezionati per chiarire eziologie specifiche, esame in cui viene prelevata, attraverso un catetere, una piccola porzione di tessuto ventricolare, che poi viene analizzata in laboratorio per una valutazione microscopica;
- test genetico, utile nei casi familiari e per lo screening dei parenti di primo grado che causano la malattia nella sua forma ereditaria.
I sintomi e le complicanze della cardiomiopatia dilatativa
I principali sintomi della cardiomiopatia dilatativa sono dovuti al quadro clinico di scompenso cardiaco, quindi:
- dispnea, sensazione di affanno che può verificarsi sia sotto sforzo fisico che a riposo;
- dolore toracico (meno frequente rispetto alla cardiopatia ischemica);
- battito cardiaco irregolare;
- maggiore affaticabilità;
- edemi periferici, dovuti all’accumulo di liquidi e che si manifestano con il gonfiore degli arti.
La cardiomiopatia dilatativa inoltre può comportare complicanze anche serie:
- insufficienza cardiaca;
- aritmie;
- eventi tromboembolici;
- morte improvvisa.
Cura e trattamento della cardiomiopatia dilatativa
In alcune forme secondarie, se la causa viene rimossa precocemente, la funzione cardiaca può migliorare parzialmente. Questo si verifica, per esempio, se la causa determinante dell’insorgenza della malattia è un fattore esterno (es. l’uso di alcol), in tal caso, infatti, la sospensione del fattore causale può favorire un recupero funzionale variabile che ha indotto la patologia per far rientrare il disturbo.
La cardiomiopatia dilatativa, dovuta ad altre cause, non è curabile, ma i sintomi e le eventuali complicanze possono essere gestiti con:
- trattamento farmacologico dello scompenso cardiaco secondo linee guida (ACE-inibitori/ARNI, beta-bloccanti, antagonisti dell’aldosterone, SGLT2-inibitori), farmaci che riducono il carico emodinamico e migliorano la funzione ventricolare, e farmaci indicati per i sintomi dello scompenso cardiaco, come beta-bloccanti e ace-inibitori, diuretici, sartani.
- impianto di un defibrillatore automatico o dispositivi di resincronizzazione cardiaca (CRT) nei pazienti selezionati, dispositivi in grado di monitorare costantemente il ritmo del battito cardiaco e, in caso di aritmia, interrompere aritmie potenzialmente letali tramite stimolazione o defibrillazione;
- trapianto cardiaco nei casi avanzati resistenti alla terapia.
Fonti
- Cardiomiopatia dilatativa. AICARM;
- Brieler J, Breeden MA, Tucker J. Cardiomyopathy: An Overview. Am Fam Physician. 2017 Nov 15;96(10):640-646. PMID: 29431384;
- Japp AG, Gulati A, Cook SA, Cowie MR, Prasad SK. The Diagnosis and Evaluation of Dilated Cardiomyopathy. J Am Coll Cardiol. 2016 Jun 28;67(25):2996-3010. doi: 10.1016/j.jacc.2016.03.590. PMID: 27339497;
- Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017 Jul 22;390(10092):400-414. doi: 10.1016/S0140-6736(16)31713-5. Epub 2017 Feb 10. PMID: 28190577;
- Halliday BP, Cleland JGF, Goldberger JJ, Prasad SK. Personalizing Risk Stratification for Sudden Death in Dilated Cardiomyopathy: The Past, Present, and Future. Circulation. 2017;136(2):215-231. doi:10.1161/CIRCULATIONAHA.116.027134
- 2023 ESC Guidelines for the management of cardiomyopathies. European Heart Journal. 2023. doi:10.1093/eurheartj/ehad194
- 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. European Heart Journal. 2021;42(36):3599–3726. doi:10.1093/eurheartj/ehab368
- Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA Guideline for the Management of Heart Failure. Journal of the American College of Cardiology. 2022;79(17):e263–e421. doi:10.1016/j.jacc.2021.12.012
- StatPearls Publishing. Dilated Cardiomyopathy. Updated 2024.